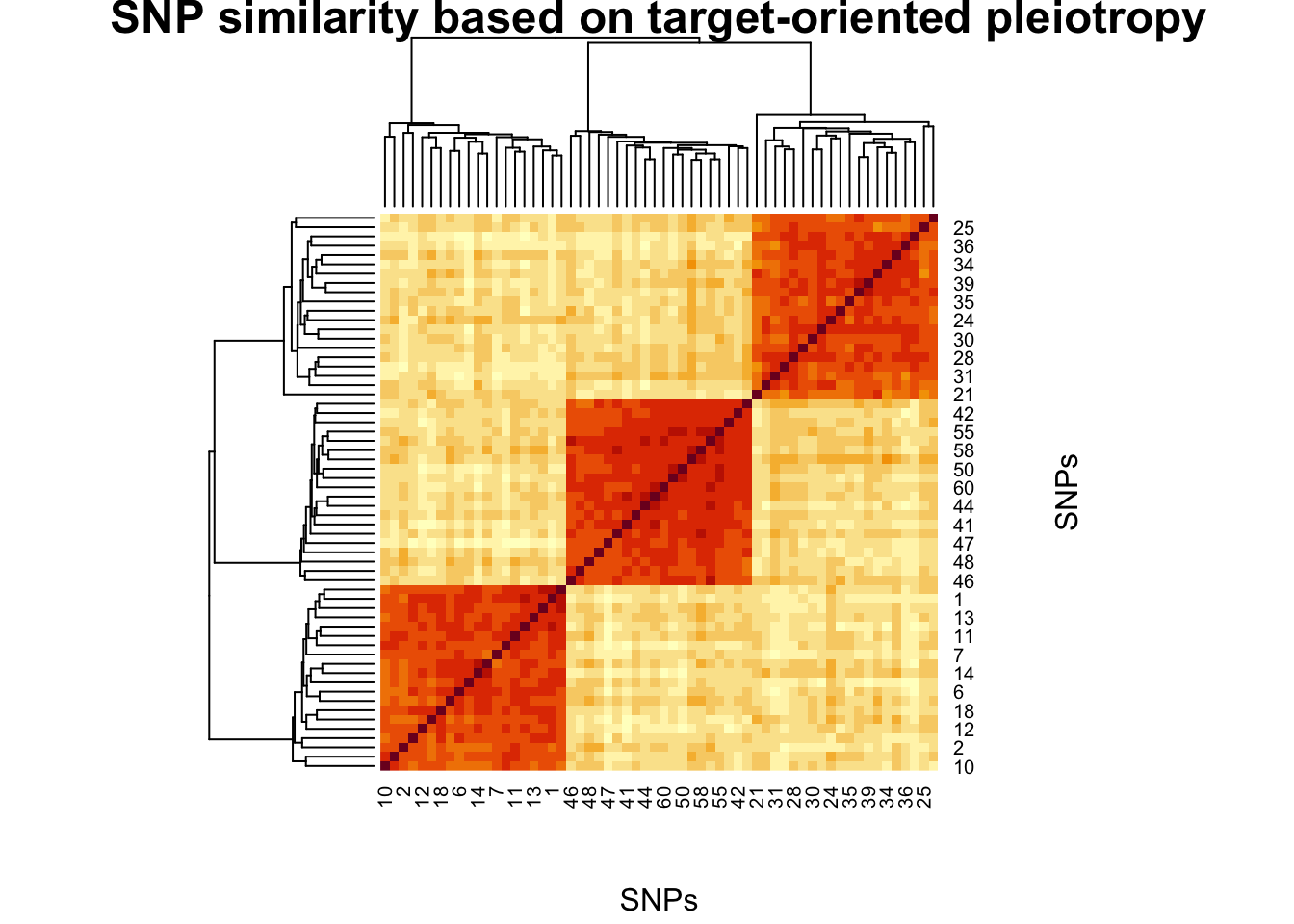
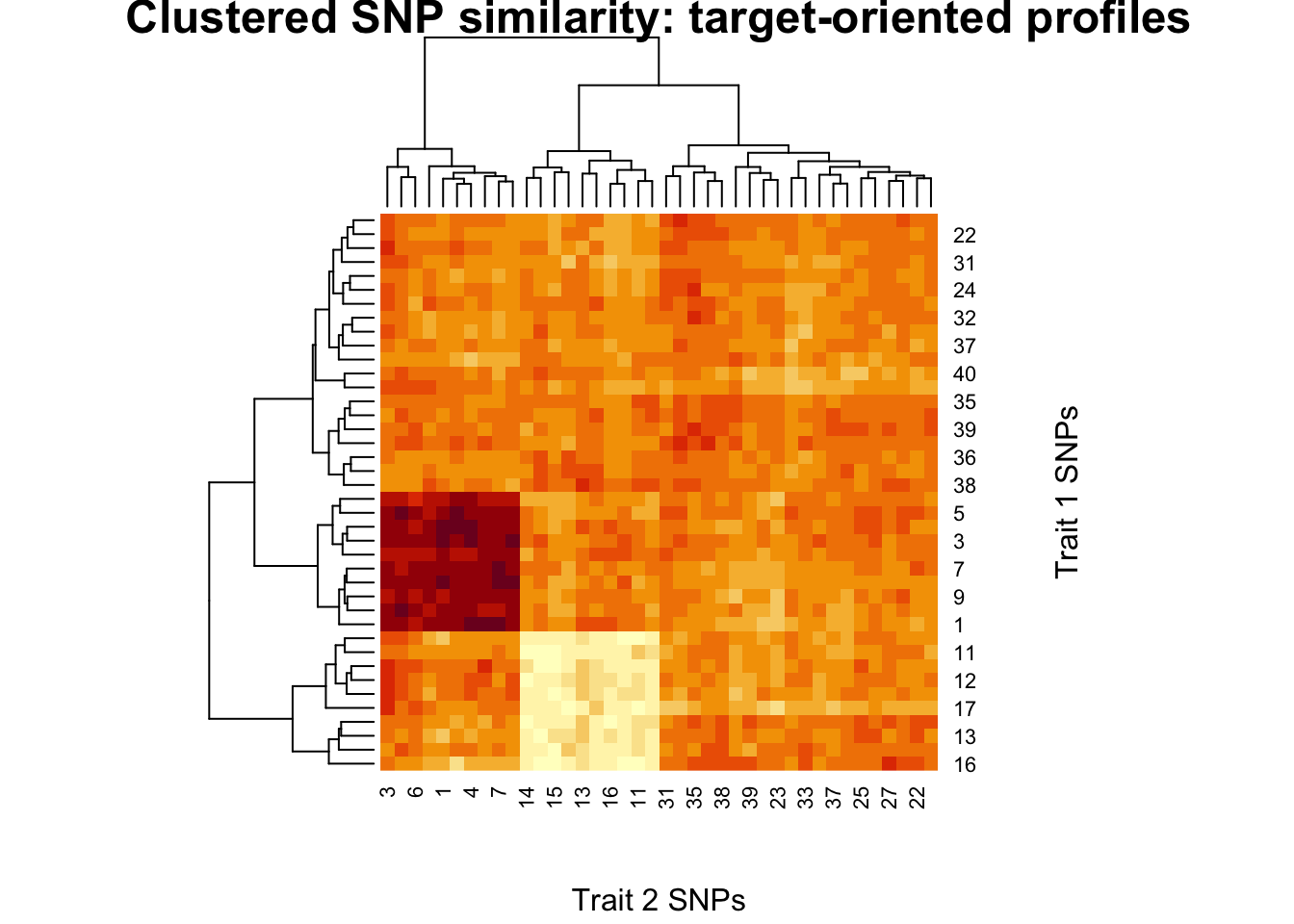
set.seed(1)
# Dimensions
n_trait <- 80 # GPMap pleiotropic traits
n1 <- 40 # SNPs for target trait 1
n2 <- 40 # SNPs for target trait 2
K <- 4 # latent biological modules
# Trait-module loadings: each module affects a subset of pleiotropic traits
B <- matrix(0, n_trait, K)
B[1:20, 1] <- 1 # shared module A
B[21:40, 2] <- 1 # shared module B
B[41:60, 3] <- 1 # trait-1-specific module
B[61:80, 4] <- 1 # trait-2-specific module
# SNP-module memberships
M1 <- matrix(0, n1, K)
M2 <- matrix(0, n2, K)
M1[1:10, 1] <- 1 # trait 1 SNPs in shared module A
M1[11:20, 2] <- 1 # trait 1 SNPs in shared module B
M1[21:40, 3] <- 1 # trait 1-specific SNPs
M2[1:10, 1] <- 1 # trait 2 SNPs in shared module A
M2[11:20, 2] <- 1 # trait 2 SNPs in shared module B
M2[21:40, 4] <- 1 # trait 2-specific SNPs
# Allow some SNPs to have opposite target-trait direction
z1 <- rnorm(n1, mean = 5, sd = 1)
z2 <- rnorm(n2, mean = 5, sd = 1)
z1[11:20] <- -z1[11:20] # module B has opposite direction for trait 1
z2[11:20] <- z2[11:20] # module B positive for trait 2
# Generate raw pleiotropy matrices
# rows = pleiotropic traits, columns = SNPs
X1 <- B %*% t(M1) + matrix(rnorm(n_trait * n1, 0, 0.5), n_trait, n1)
X2 <- B %*% t(M2) + matrix(rnorm(n_trait * n2, 0, 0.5), n_trait, n2)
# Target-oriented profiles
X1_star <- sweep(X1, 2, z1, `*`)
X2_star <- sweep(X2, 2, z2, `*`)
# Cross-trait similarity: SNPs from trait 1 vs SNPs from trait 2
cosine_sim <- function(A, B) {
A_norm <- sweep(A, 2, sqrt(colSums(A^2)), `/`)
B_norm <- sweep(B, 2, sqrt(colSums(B^2)), `/`)
t(A_norm) %*% B_norm
}
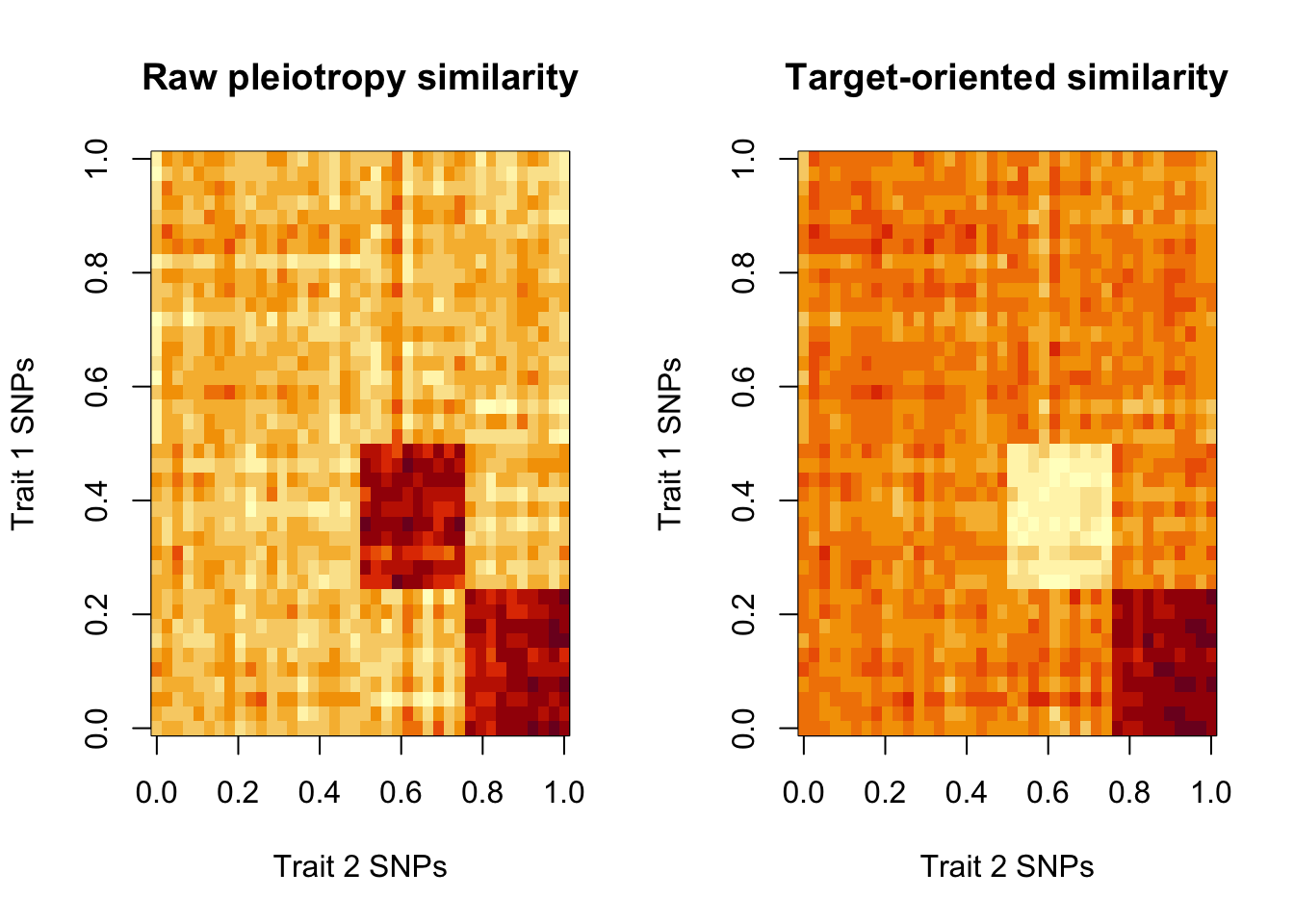
S_raw <- cosine_sim(X1, X2)
S_star <- cosine_sim(X1_star, X2_star)
# Visualise
par(mfrow = c(1, 2))
image(
S_raw[nrow(S_raw):1, ],
main = "Raw pleiotropy similarity",
xlab = "Trait 2 SNPs",
ylab = "Trait 1 SNPs"
)
image(
S_star[nrow(S_star):1, ],
main = "Target-oriented similarity",
xlab = "Trait 2 SNPs",
ylab = "Trait 1 SNPs"
)