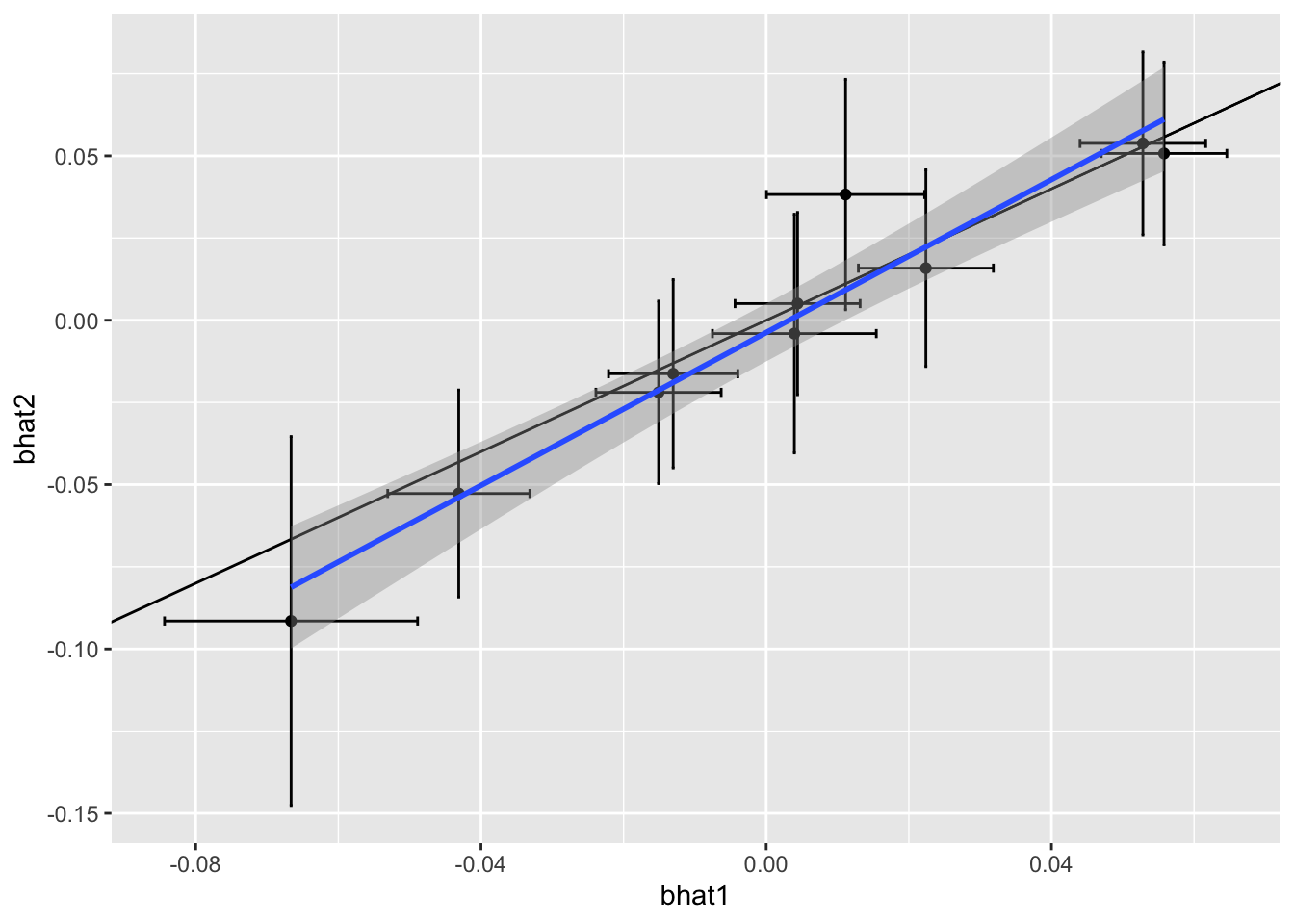
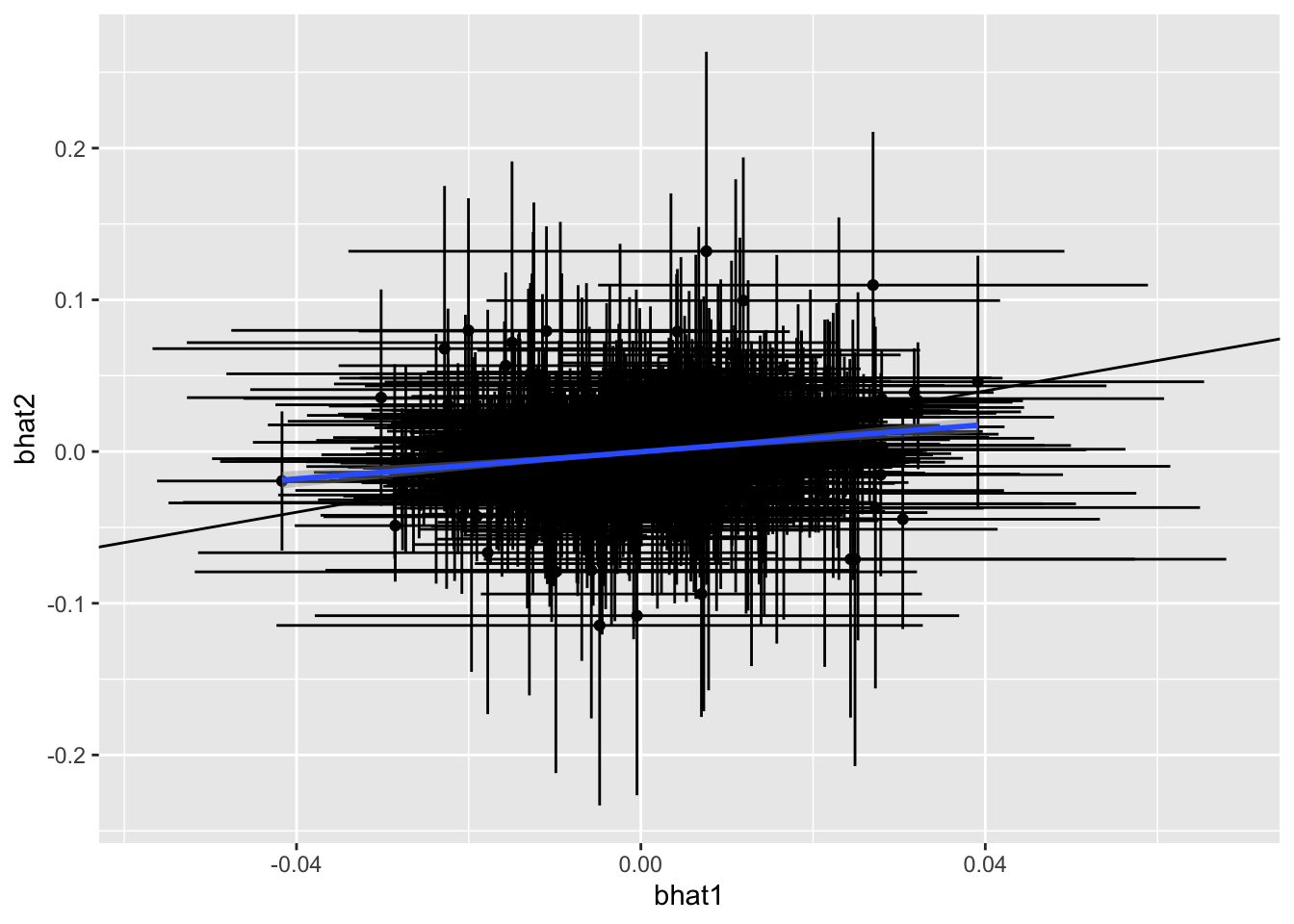
We can calculate the expected number to replicate given the differential power (due to sample size and allele frequency differences across studies), and compare this to the observed number to replicate. If fewer replicate than expected, then this is evidence of heterogeneity in the effect sizes.
#' Estimate expected vs observed replication of effects between discovery and replication datasets#' #' Taken from Okbay et al 2016. Under the assumption that all discovery effects are unbiased, what fraction of associations would replicate in the replication dataset, given the differential power of the discovery and replication datasets.#' Uses standard error of the replication dataset to account for differences in sample size and distribution of independent variable#' #' @param b_disc Vector of discovery betas#' @param b_rep Vector of replication betas#' @param se_disc Vector of discovery standard errors#' @param se_rep Vector of replication standard errors#' @param alpha Nominal replication significance threshold#' #' @return List of results#' - res: aggregate expected replication rate vs observed replication rate#' - variants: per variant expected replication ratesprop_overlap <-function(b_disc, b_rep, se_disc, se_rep, alpha) { p_sign <-pnorm(-abs(b_disc) / se_disc) *pnorm(-abs(b_disc) / se_rep) + ((1-pnorm(-abs(b_disc) / se_disc)) * (1-pnorm(-abs(b_disc) / se_rep))) p_sig <-pnorm(-abs(b_disc) / se_rep +qnorm(alpha /2)) + (1-pnorm(-abs(b_disc) / se_rep -qnorm(alpha /2))) p_rep <-pnorm(abs(b_rep) / se_rep, lower.tail =FALSE) res <- tibble::tibble(nsnp =length(b_disc),metric =c("Sign", "Sign", "P-value", "P-value"),datum =c("Expected", "Observed", "Expected", "Observed"),value =c(sum(p_sign, na.rm =TRUE), sum(sign(b_disc) ==sign(b_rep)), sum(p_sig, na.rm =TRUE), sum(p_rep < alpha, na.rm =TRUE)) ) %>% dplyr::group_by(metric) %>% dplyr::do({ x <- .if(.$nsnp[1] >0) { bt <-binom.test(x=.$value[.$datum =="Observed"], n=.$nsnp[1], p=.$value[.$datum =="Expected"] / .$nsnp[1] )$p.value x$pdiff <- bt } x })return(list(res = res, variants = dplyr::tibble(sig = p_sig, sign = p_sign, )))}
This shows that we don’t expect all significant discovery associations to replicate, and indeed they don’t, but the rate of observed replication matches the expected rate of replication. The pdiff column is the p-value from a binomial test comparing the observed and expected replication rates, a low p-value indicates that the observed replication rate is substantially different from the expected replication rate.
Heterogeneity
Evaluate if any one particular variant has heterogeneity in effect sizes across the two studies. This is done using Cochrane’s Q statistic, which accounts for difference in power (based on SE) across the studies.
In this situation the effects have the same magnitude (so the p-values should give comparable estimates), but the signs are different for some of the effects. This is reflected in the observed replication rate for the sign, which is lower than the expected replication rate.
Note that trying to replicate discovery SNPs can have problems because of winner’s curse in the discovery estimate, which could lead to lower observed replication rates than expected even accounting for differences in power.
Summary
The regression of effects across studies might not be 1 even when the true effects are consistent across studies, because power differences can distort the relationship
Estimating the observed vs expected replication rates can help to identify if there are systematic differences in effect sizes across studies
Estimating the per-variant heterogeneity can estimate if there are any particular variants that have different effect sizes across studies