A <- matrix(c(1.54, 35, -1, 5), nrow=2, byrow=T)
b <- c(4.9, 0)
prev <- solve(A, b)
prev[1] 0.5737705 0.1147541T2D rates are ~5x higher amongst South Asians than native British populations in Britain. Most likely due to environmental differences, but could it hypothetically be explained by differences in genetic architecture?
Assume polygenic common variant distribution and a disease liability threshold model.
Simultaneous equations:
\[ 4.9 = 1.54 x + 35y \]
where \(x\) is the prevalence amongst South Asians and \(y\) is the prevalence amongst white British
Also
\[ x = y * 5 \]
Calculate prevalences:
A <- matrix(c(1.54, 35, -1, 5), nrow=2, byrow=T)
b <- c(4.9, 0)
prev <- solve(A, b)
prev[1] 0.5737705 0.1147541Assume threshold is the same in both populations. Establish a threshold based on European genetic architecture
library(ggplot2)
library(dplyr)
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionsim1 <- function(mg, vg, me, ve, prev) {
n <- 100000
g <- rnorm(n, mg, sqrt(vg))
e <- rnorm(n, me, sqrt(ve))
l <- g + e
thresh <- quantile(l, 1-prev)
return(thresh)
}
thresh <- sim1(0, 0.4, 0, 0.6, prev[2])
thresh88.52459%
1.204965 Estimate prevalence given a threshold when parameters change
sim2 <- function(mg, vg, me, ve, thresh) {
n <- 100000
g <- rnorm(n, mg, sqrt(vg))
e <- rnorm(n, me, sqrt(ve))
l <- g + e
prev <- sum(l > thresh) / n
return(prev)
}
sim2(0.1, 0.4, 0, 0.6, thresh)[1] 0.13381Enumerate possibilities
param <- expand.grid(
mg = seq(0, 4, by=0.01),
vg = 0.4,
me = 0,
ve = 0.6,
thresh = thresh,
prev = NA
)
for(i in 1:nrow(param)) {
param$prev[i] <- sim2(param$mg[i], param$vg[i], param$me[i], param$ve[i], param$thresh[i])
}
param$ratio = param$prev / prev[2]
ggplot(param, aes(x=mg, y=ratio)) +
geom_line() +
geom_hline(yintercept=5, linetype="dotted")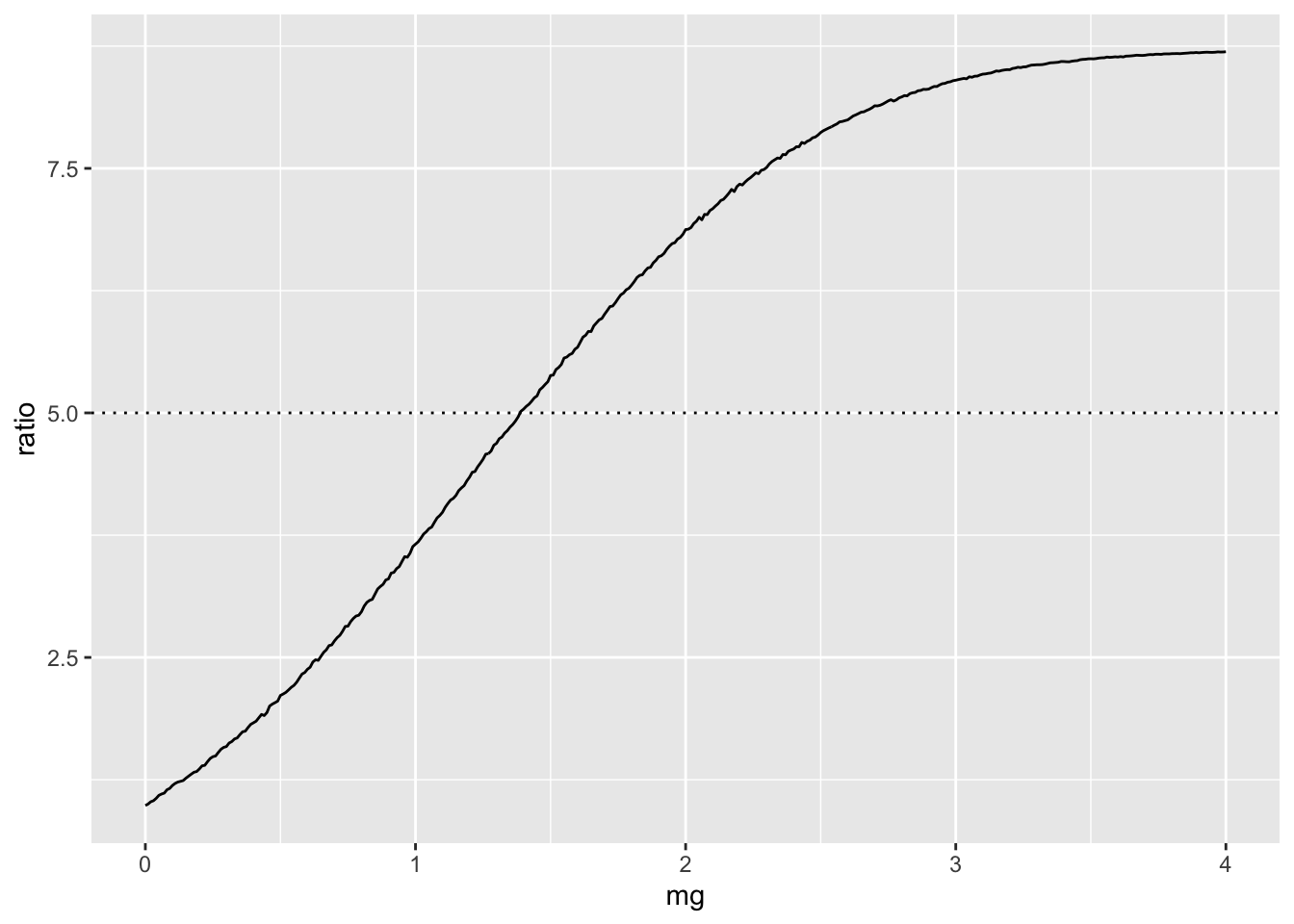
This shows how much larger the mean of the genetic liability would have to be to lead to 5x prevalence
Comparison of distributions:
mg <- param$mg[which(param$ratio >= 5)[1]]
bind_rows(
tibble(pop="EUR", x=seq(-3, 3, length.out=1000), val = dnorm(x, sd=sqrt(0.4))),
tibble(pop="SAS", x=seq(-3+mg, 3+mg, length.out=1000), val = dnorm(x, m=mg, sd=sqrt(0.4)))
) %>% ggplot(., aes(x=x, y=val)) +
geom_line(aes(colour=pop)) +
geom_vline(xintercept=thresh)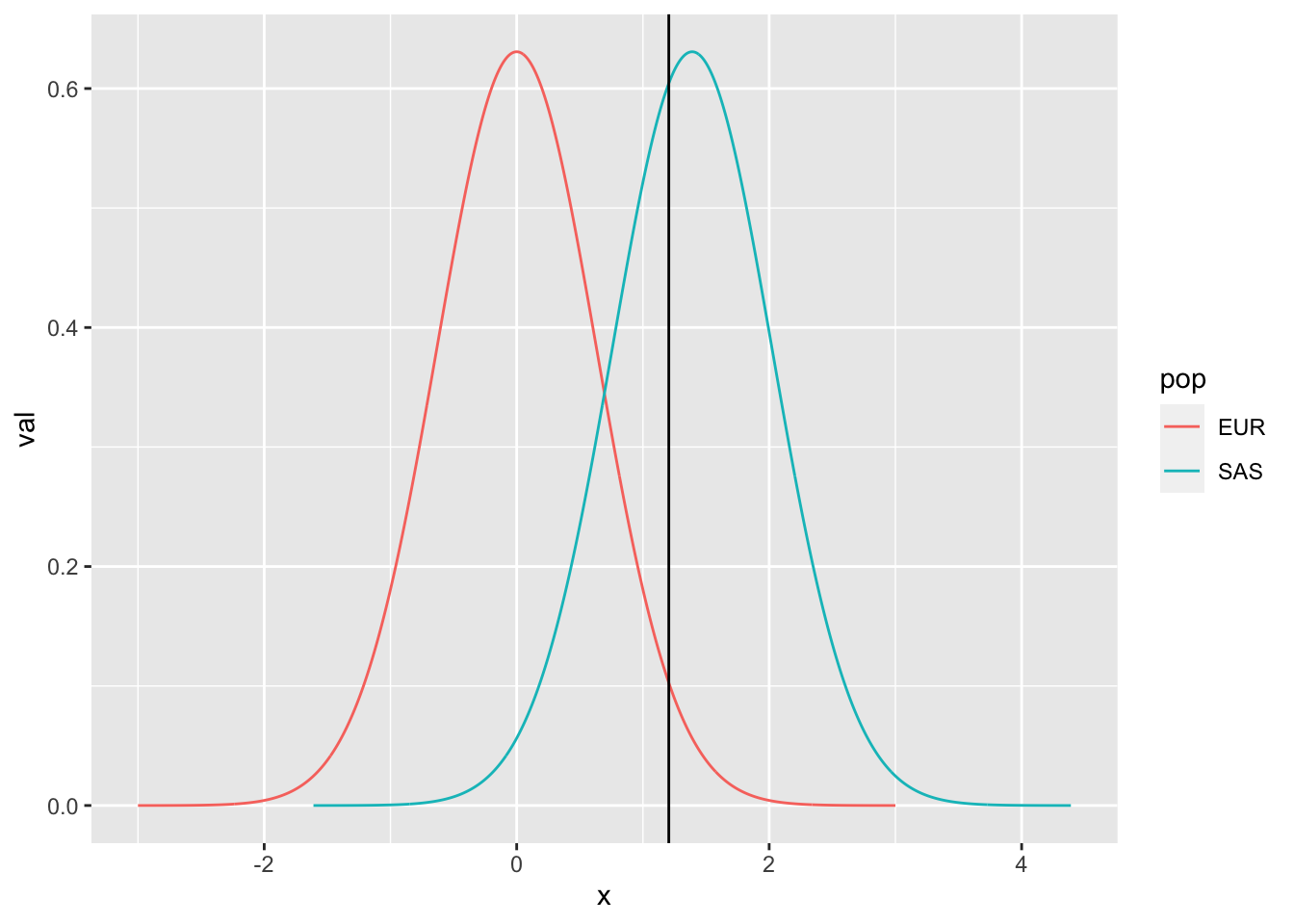
Is it plausible in terms of just different allele frequencies?
nsnp <- 10000
af <- rbeta(nsnp, 1, 1) / 2
hist(af)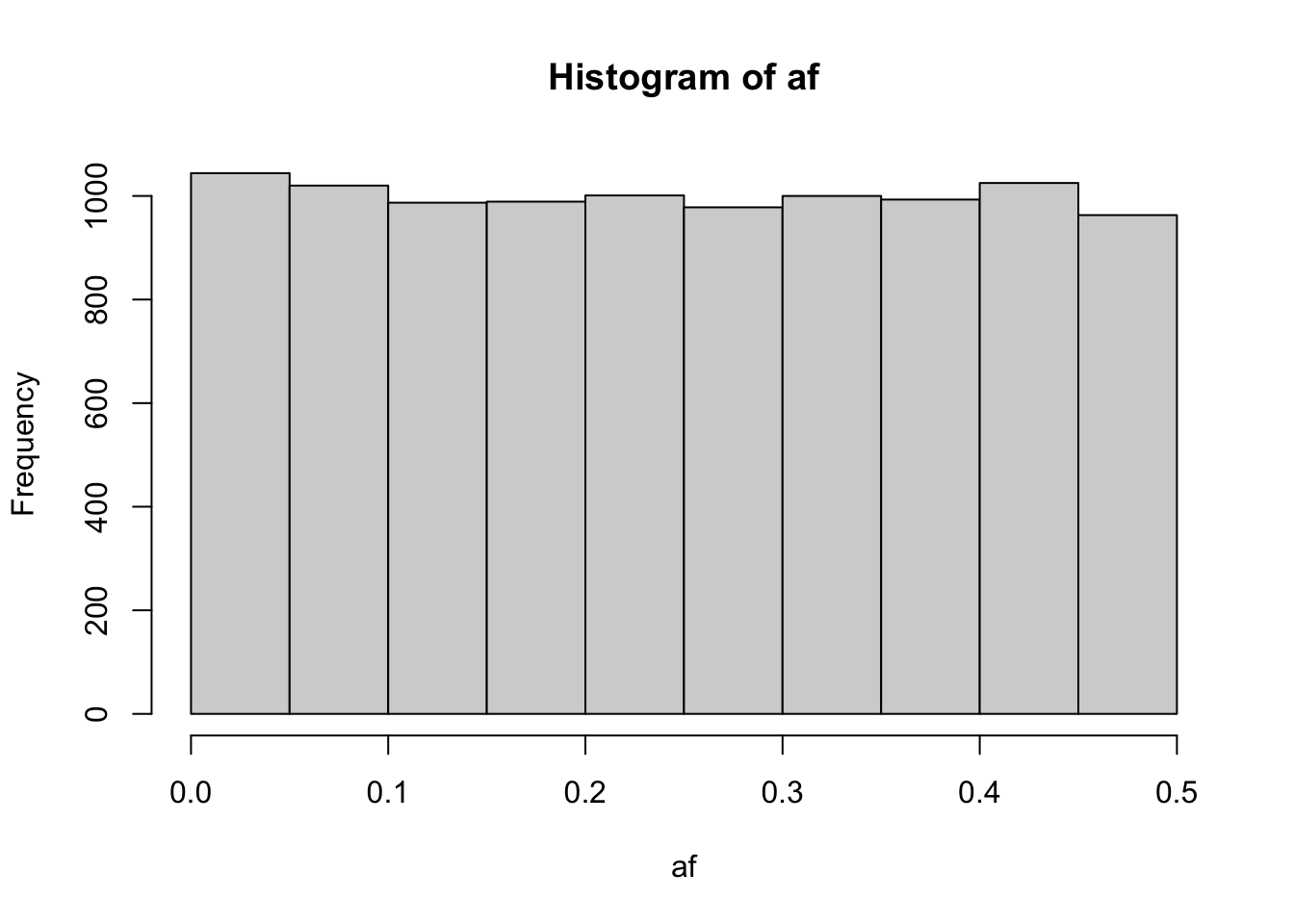
b <- rnorm(nsnp, 0, (2*af*(1-af))^-0.4)
vg <- sum(b^2 * 2 * af * (1-af))
b <- b / sqrt(vg) * 0.4
plot(b ~ af)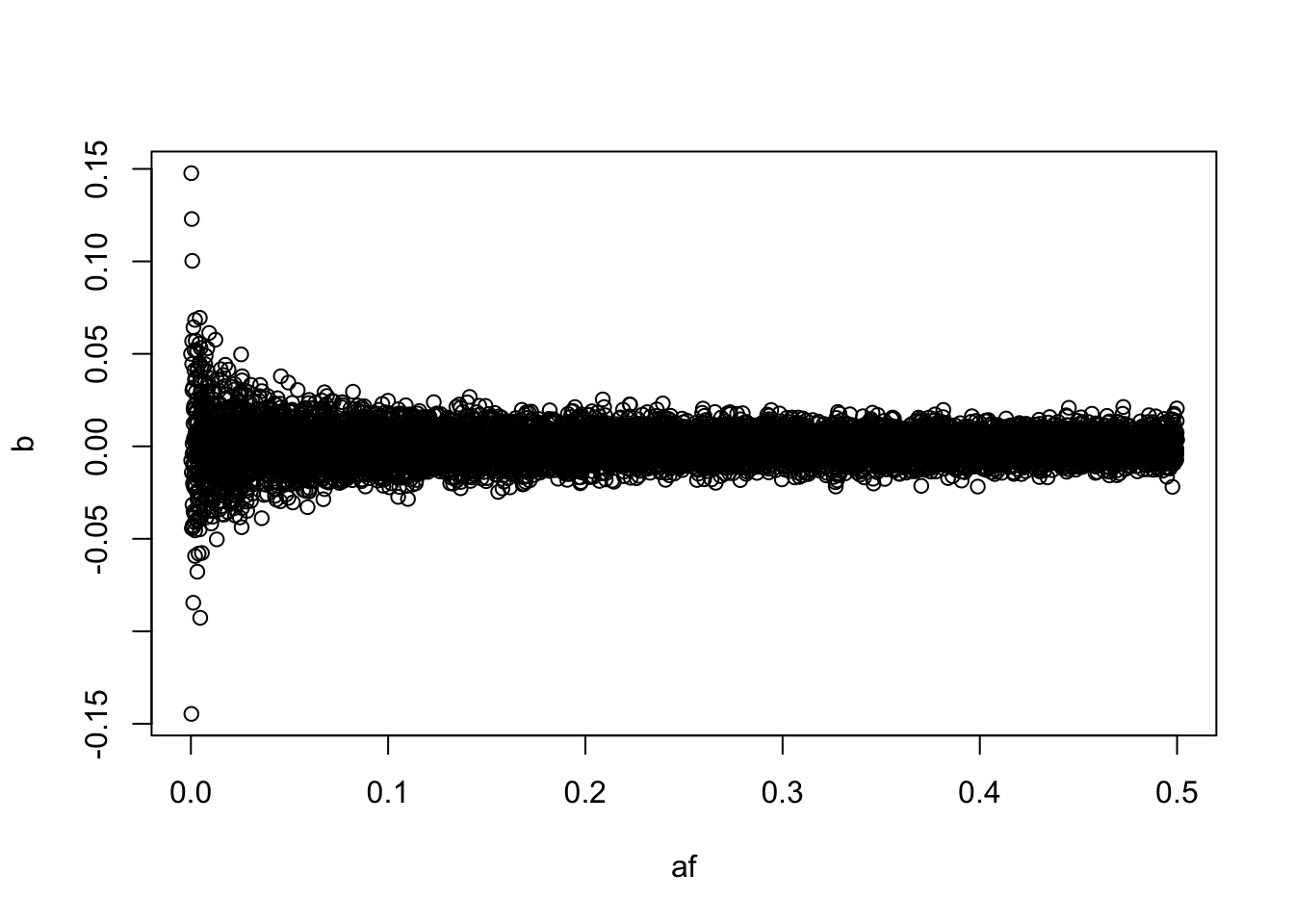
nid <- 1000
g1 <- sapply(af, \(x) { rbinom(nid, 2, x) })
score1 <- g1 %*% b
hist(score1)
sd(score1)[1] 0.383443sessionInfo()R version 4.3.2 (2023-10-31)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Ventura 13.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_GB.UTF-8/en_GB.UTF-8/en_GB.UTF-8/C/en_GB.UTF-8/en_GB.UTF-8
time zone: Europe/London
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] dplyr_1.1.4 ggplot2_3.4.2
loaded via a namespace (and not attached):
[1] vctrs_0.6.4 cli_3.6.1 knitr_1.45 rlang_1.1.2
[5] xfun_0.41 generics_0.1.3 jsonlite_1.8.7 labeling_0.4.2
[9] glue_1.6.2 colorspace_2.1-0 htmltools_0.5.7 scales_1.2.1
[13] fansi_1.0.5 rmarkdown_2.25 grid_4.3.2 evaluate_0.23
[17] munsell_0.5.0 tibble_3.2.1 fastmap_1.1.1 yaml_2.3.7
[21] lifecycle_1.0.4 compiler_4.3.2 htmlwidgets_1.6.3 pkgconfig_2.0.3
[25] farver_2.1.1 digest_0.6.33 R6_2.5.1 tidyselect_1.2.0
[29] utf8_1.2.4 pillar_1.9.0 magrittr_2.0.3 withr_2.5.2
[33] tools_4.3.2 gtable_0.3.3