set.seed(1234)
n <- 10000
u <- rnorm(n)
x <- rnorm(n) + u
a <- rnorm(n) + x + u - 1
y <- rbinom(n, 1, plogis(a))
table(y)y
0 1
6331 3669 Using REML to adjust for pedigree and then taking residuals to estimate PRS association. However the original trait is binary and the residuals are continuous, how to interpret the effect size?
Simulate data - a confounder influences x and y, x influences y, and y is binary.
set.seed(1234)
n <- 10000
u <- rnorm(n)
x <- rnorm(n) + u
a <- rnorm(n) + x + u - 1
y <- rbinom(n, 1, plogis(a))
table(y)y
0 1
6331 3669 Estimate using glm
summary(glm(y ~ x + u, family="binomial"))$coef Estimate Std. Error z value Pr(>|z|)
(Intercept) -0.8456496 0.02749282 -30.75892 9.290785e-208
x 0.7864091 0.02827687 27.81104 3.189961e-170
u 0.8236558 0.03882063 21.21696 6.659380e-100Transformation term
mu <- sum(y) / length(y)
tr <- mu * (1-mu)Get residuals (raw)
yres_raw <- residuals(glm(y ~ u, family="binomial"), type="response")
summary(lm(yres_raw ~ x))$coef Estimate Std. Error t value Pr(>|t|)
(Intercept) 9.859144e-05 0.004038968 0.02441006 9.805260e-01
x 6.044366e-02 0.002850011 21.20821889 1.114094e-97After transformation
lm(yres_raw ~ x)$coef[2] / tr x
0.260214 Get residuals (deviance)
yres_dev <- residuals(glm(y ~ u, family="binomial"))
summary(lm(yres_dev ~ x))$coef Estimate Std. Error t value Pr(>|t|)
(Intercept) -0.06563045 0.009654231 -6.798102 1.120585e-11
x 0.20379274 0.006812301 29.915404 2.104826e-188After transformation
lm(yres_dev ~ x)$coef[2] / tr x
0.8773415 Range
library(dplyr)
Attaching package: 'dplyr'The following objects are masked from 'package:stats':
filter, lagThe following objects are masked from 'package:base':
intersect, setdiff, setequal, unionparam <- expand.grid(
b=seq(-1, 1, by=0.1),
int=seq(-2,0, by=0.2)
)
o <- lapply(1:nrow(param), \(i) {
u <- rnorm(n)
x <- rnorm(n) + u
a <- rnorm(n) + x * param$b[i] + u + param$int[i]
y <- rbinom(n, 1, plogis(a))
mu <- sum(y) / length(y)
tr <- mu * (1-mu)
bhat <- glm(y ~ x + u, family="binomial")$coef[2]
yres_raw <- residuals(glm(y ~ u, family="binomial"), type="response")
bhat_raw <- lm(yres_raw ~ x)$coef[2]
yres_dev <- residuals(glm(y ~ u, family="binomial"))
bhat_dev <- lm(yres_dev ~ x)$coef[2]
tibble(bhat, bhat_raw, bhat_dev, bhat_raw_tr = bhat_raw / tr, bhat_dev_tr=bhat_dev / tr, tr=tr)
}) %>% bind_rows() %>% bind_cols(param, .)plot
pairs(o %>% select(-c(int, tr)))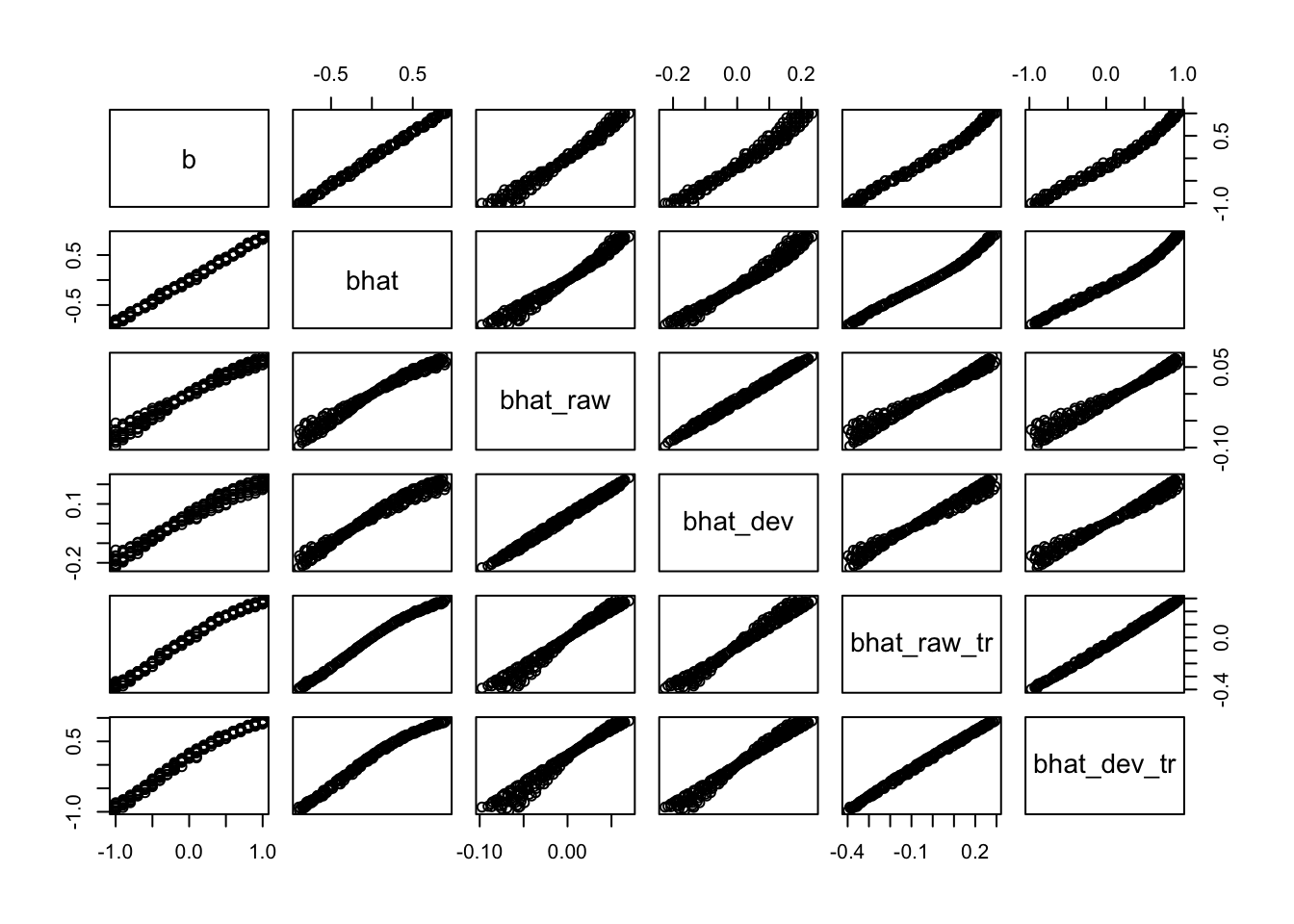
dim(o)[1] 231 8Looks like the deviance residuals are on the same scale after transformation, but will need a bit of work to translate between deviance and raw
https://data.library.virginia.edu/understanding-deviance-residuals/
sessionInfo()R version 4.3.0 (2023-04-21)
Platform: aarch64-apple-darwin20 (64-bit)
Running under: macOS Monterey 12.6.8
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Europe/London
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] dplyr_1.1.2
loaded via a namespace (and not attached):
[1] digest_0.6.31 utf8_1.2.3 R6_2.5.1 fastmap_1.1.1
[5] tidyselect_1.2.0 xfun_0.39 magrittr_2.0.3 glue_1.6.2
[9] tibble_3.2.1 knitr_1.43 pkgconfig_2.0.3 htmltools_0.5.5
[13] generics_0.1.3 rmarkdown_2.22 lifecycle_1.0.3 cli_3.6.1
[17] fansi_1.0.4 vctrs_0.6.2 withr_2.5.0 compiler_4.3.0
[21] rstudioapi_0.14 tools_4.3.0 pillar_1.9.0 evaluate_0.21
[25] yaml_2.3.7 rlang_1.1.1 jsonlite_1.8.5 htmlwidgets_1.6.2