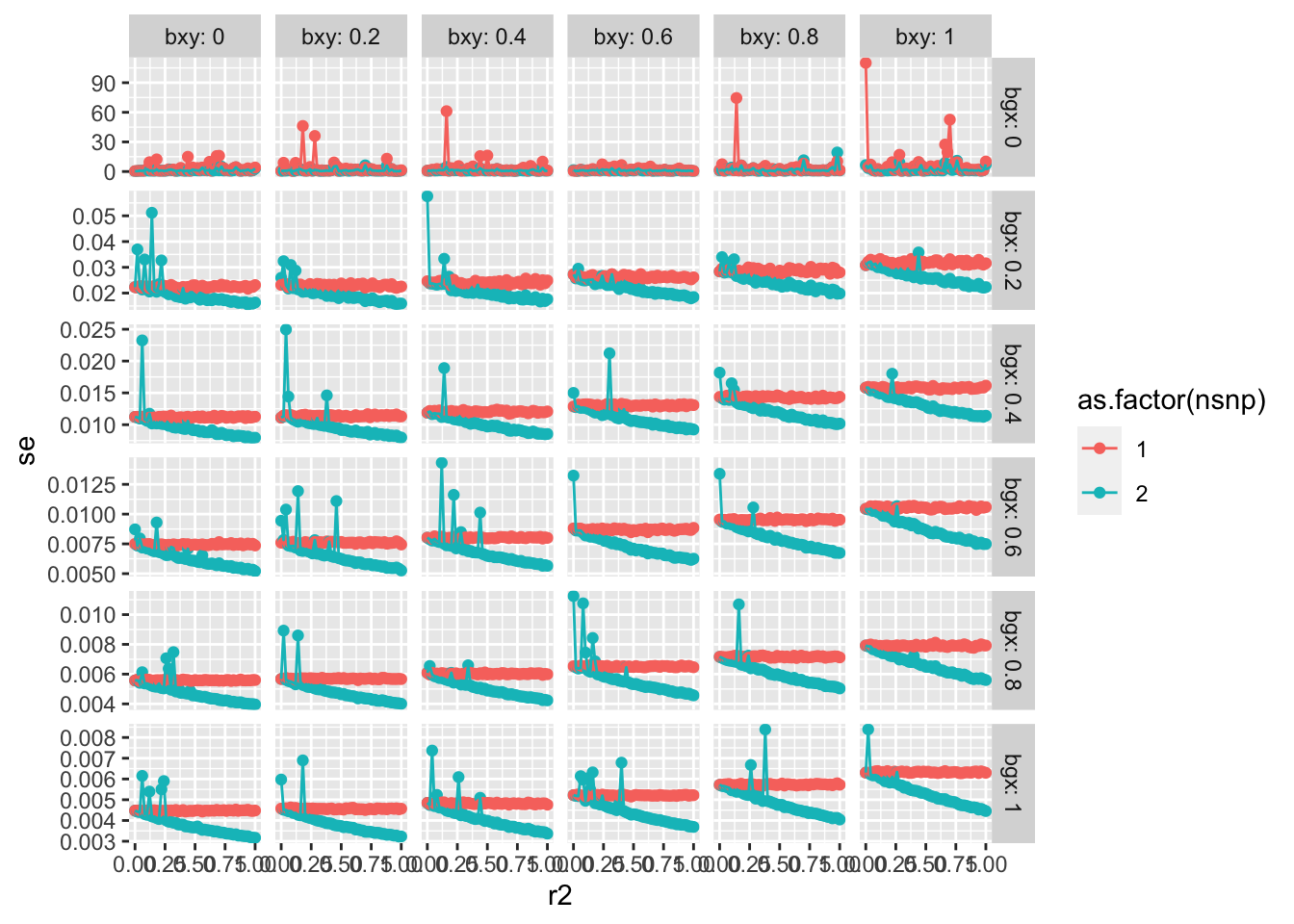
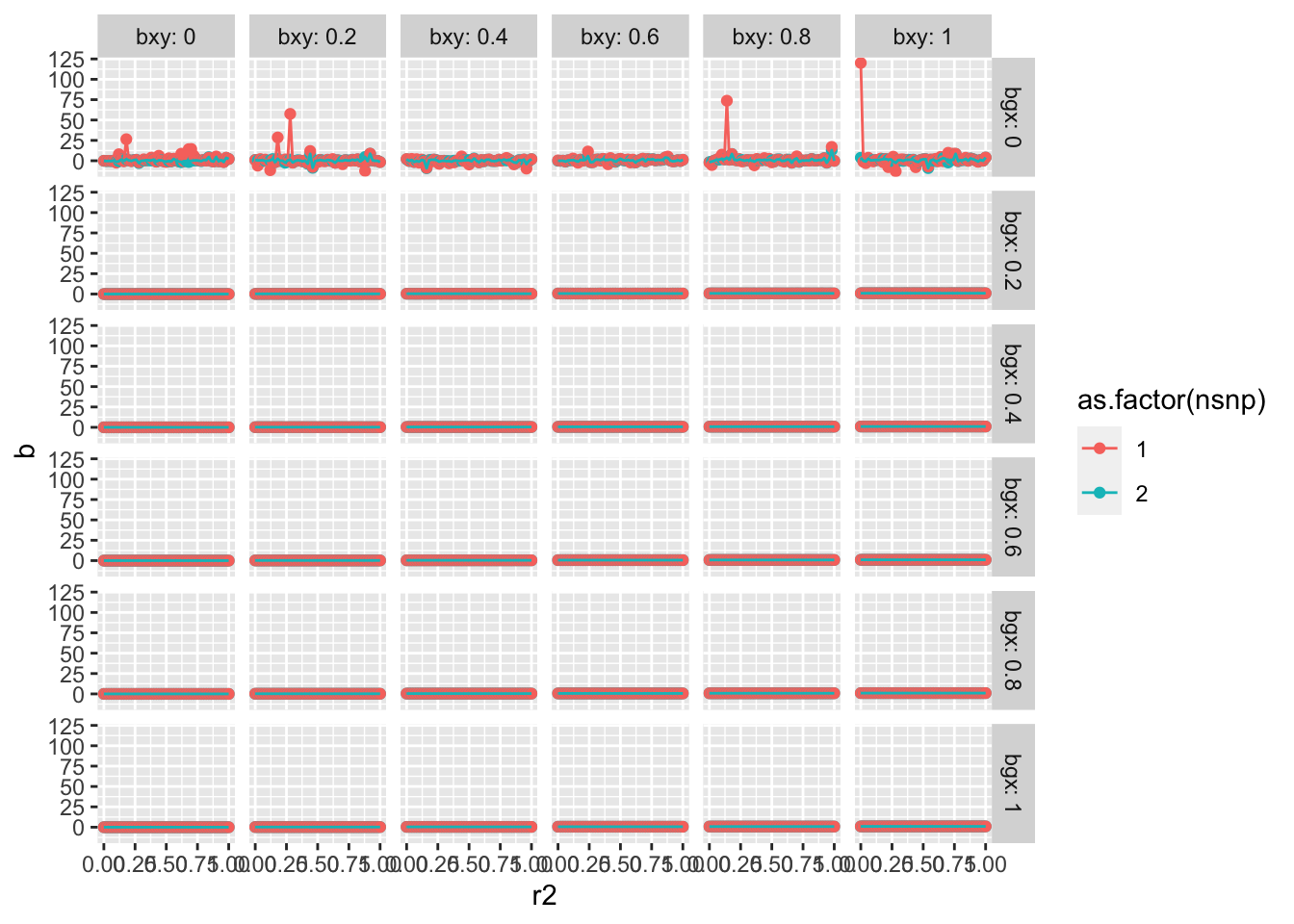
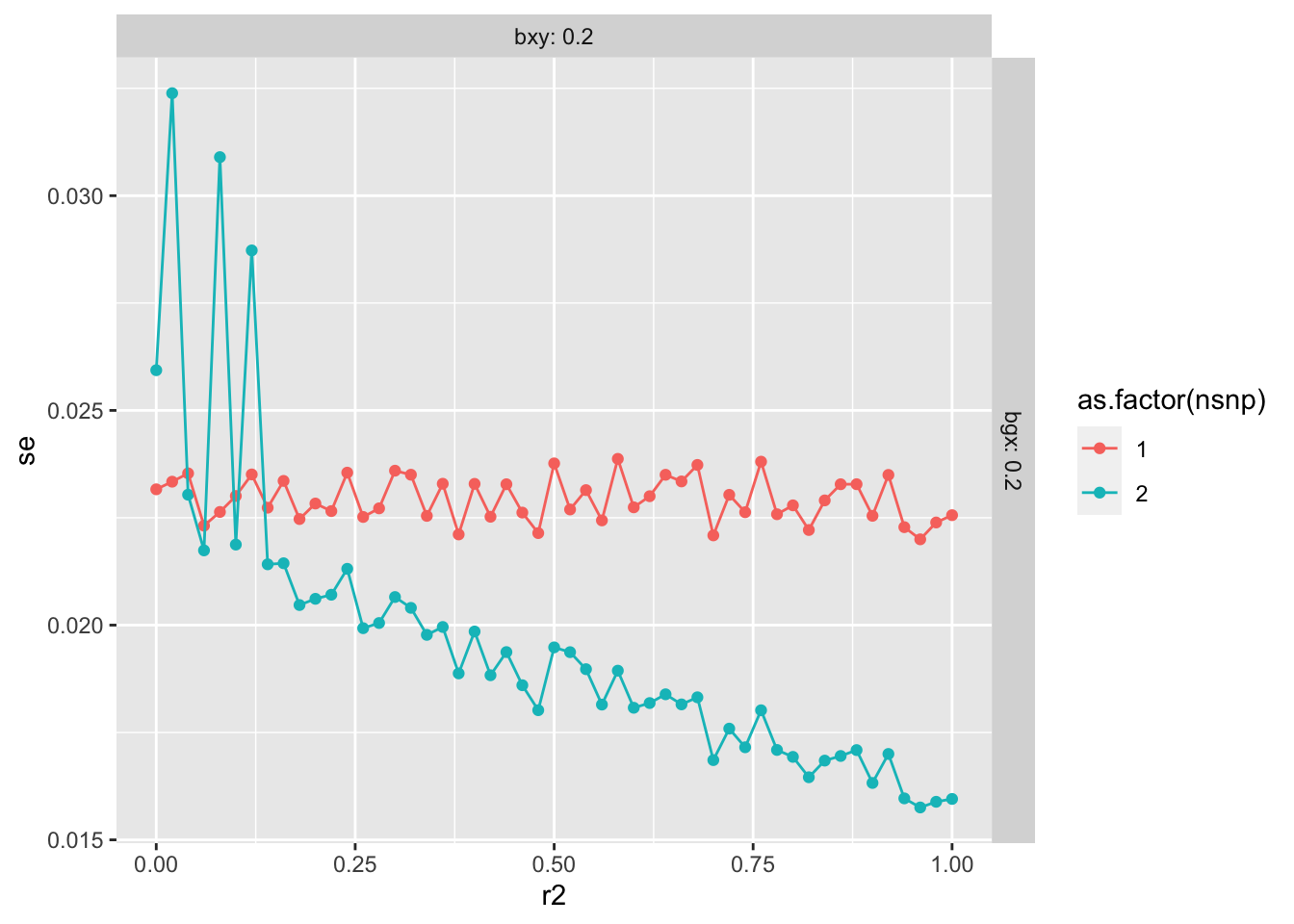
4000 instruments for educational attainment using clumping r2 = 0.1, and se doubles when using r2 = 0.001.
That smaller standard error is either due to the R2 in the exposure being higher or the non-independence of effects artificially increasing precision, or a mixture of both.
So the question is the impact of the latter – if we have some true correlation structure with realistic F stats at a specific locus, and then we try to clump at r2 = 0.001 vs 0.1, how many instruments do we retain (it should be 1) and if more than 1, what is that impact on the standard error
library(dplyr)
Attaching package: 'dplyr'
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
library(simulateGP)library(TwoSampleMR)
TwoSampleMR version 0.5.7
[>] New: Option to use non-European LD reference panels for clumping etc
[>] Some studies temporarily quarantined to verify effect allele
[>] See news(package='TwoSampleMR') and https://gwas.mrcieu.ac.uk for further details
Attaching package: 'TwoSampleMR'
The following objects are masked from 'package:simulateGP':
allele_frequency, contingency, get_population_allele_frequency
library(purrr)library(ggplot2)
Simulate causal snp g + another that is correlated with it
n <-100000g <-correlated_binomial(n, p1=0.5, p2=0.5, rho=sqrt(0.1))# x caused by just snp 1x <- g[,1] *0.5+rnorm(n)y <- x *0.5+rnorm(n)# MR using both SNPs, treating as if they are independentget_effs(x, y, g) %>%mr(method="mr_ivw") %>% str
Analysing 'X' on 'Y'
'data.frame': 1 obs. of 9 variables:
$ id.exposure: chr "X"
$ id.outcome : chr "Y"
$ outcome : chr "Y"
$ exposure : chr "X"
$ method : chr "Inverse variance weighted"
$ nsnp : int 2
$ b : num 0.497
$ se : num 0.00956
$ pval : num 0
# MR using just the causal SNPget_effs(x, y, g[,1, drop=F]) %>%mr(method=c("mr_ivw", "mr_wald_ratio")) %>% str
Analysing 'X' on 'Y'
'data.frame': 1 obs. of 9 variables:
$ id.exposure: chr "X"
$ id.outcome : chr "Y"
$ outcome : chr "Y"
$ exposure : chr "X"
$ method : chr "Wald ratio"
$ nsnp : num 1
$ b : num 0.496
$ se : num 0.0101
$ pval : num 0
There’s hardly any difference in the SE here. Try over a range of scenarios
res <-map(1:nrow(param), \(i){ g <-correlated_binomial(param$n[i], p1=0.5, p2=0.5, rho=sqrt(param$r2[i]))# x caused by just snp 1 x <- g[,1] * param$bgx[i] +rnorm(n) y <- x * param$bxy[i] +rnorm(n)bind_rows(get_effs(x, y, g) %>% {suppressMessages(mr(., method="mr_ivw"))},get_effs(x, y, g[,1, drop=F]) %>% {suppressMessages(mr(., method="mr_wald_ratio"))} ) %>%mutate(sim=param$sim[i]) %>%return()}) %>% bind_rows %>%inner_join(param, ., by="sim")
Warning in summary.lm(stats::lm(b_out ~ -1 + b_exp, weights = 1/se_out^2)):
essentially perfect fit: summary may be unreliable
Relaxed r2 e.g. from 0 to 0.1 doesn’t seem to have a huge impact on standard errors
In the one SNP situation relaxed r2 has no impact on bias, and could only plausibly change things under substantial heterogeneity which correlates with overrepresentation.
More realistic simulations would look at whether this changes when the p-value at the second locus is very large, and would also look at the probability of erroneously keeping multiple loci for a single causal variant