In a previous post looked at the rate of bias as GWAS sample size increases. It was predicated on smaller effects more likely acting via confounders.
What is the justification for smaller effects acting via confounders? After all, all effects likely act via mediators. Some of those mediators will confound a X-Y relationship while some will not. Is there any reason to believe that as effects get smaller they’re more likely to act via a confounder?
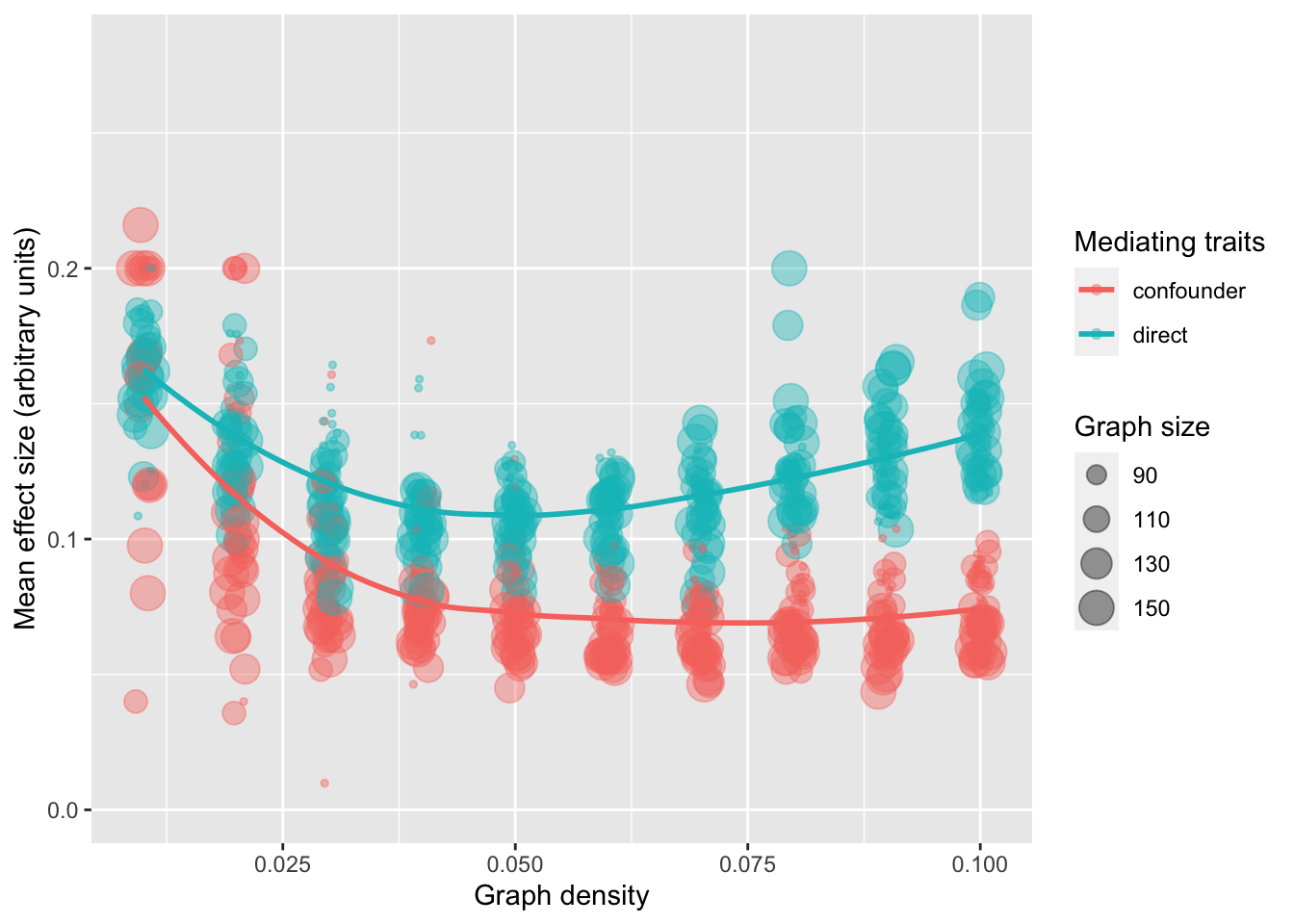
Imagine a large DAG representing the genotype-phenotype map. All nodes that have no parents are genetic variants. All other nodes are traits. If you choose any two traits, one as the exposure and one as the outcome, what is the nature of the instruments for the exposure? Hypothesis: the instruments that are mediated by confounders are likely to be more distal from the exposure, compared to those that act via non-confounding mediators.
These simulations aim to test that.
library(dagitty)library(dplyr)
Attaching package: 'dplyr'
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
library(furrr)
Loading required package: future
library(ggplot2)library(tictoc)simulategraphconf <-function(n, p) {# Generate graph g <- dagitty::randomDAG(n, p)# All ancestors anc <-lapply(names(g), \(x) ancestors(g, x))names(anc) <-names(g)# Identify genetic factors (no ancestors) temp <-lapply(anc, length) temp <-tibble(node=names(temp), nanc=unlist(temp)) gen <-subset(temp, nanc==1)$nodemessage("Number of genetic variants: ", length(gen)) traits <-subset(temp, nanc !=1)$nodemessage("Number of traits: ", length(traits))# Find distance of all genetic variants to # Find all trait pairs tp <-lapply(traits, \(tr) { temp <-ancestors(g, tr)tibble(x=temp, y=tr) %>%filter(! x %in%c(gen, tr)) }) %>% bind_rows# tp <- expand.grid(x=traits, y=traits) %>%# as_tibble() %>%# filter(x != y)message("Number of trait pairs: ", nrow(tp)) res <- furrr::future_map(1:min(nrow(tp), 500), \(i) { x_ancestors <-ancestors(g, tp$x[i]) x_ancestors <- x_ancestors[x_ancestors %in% gen] y_ancestors <-ancestors(g, tp$y[i]) y_ancestors <- y_ancestors[y_ancestors %in% gen] conf <-c() nonconf <-c()for(a in y_ancestors) { x <-paths(g, a, tp$y[i], dir=T)$pathsif(any(!grepl(paste0(" ", tp$x[i], " "), x))) { conf <-c(conf, a) } else { nonconf <-c(nonconf, a) } }bind_rows(lapply(conf, function(co) { pa <-paths(g, co, tp$x[i], directed=TRUE)$pathsif(length(pa) !=0) {tibble(x=tp$x[i],y=tp$y[i],gen=co,type="confounder",w=pa %>%sapply(., function(x) { stringr::str_count(x, "->") %>%unlist() %>% {0.2^.} }) %>% sum ) } else {NULL } }) %>%bind_rows(),lapply(nonconf, function(co) { pa <-paths(g, co, tp$x[i], directed=TRUE)$pathsif(length(pa) !=0) {tibble(x=tp$x[i],y=tp$y[i],gen=co,type="direct",w=pa %>%sapply(., function(x) { stringr::str_count(x, "->") %>%unlist() %>% {0.2^.} }) %>% sum ) } else {NULL } }) %>%bind_rows() ) }) %>%bind_rows()# res$causal <- paste(res$x, res$y) %in% paste(tpc$x, tpc$y)return(res)}set.seed(1234) # note this doesn't work for dagitty - for seed should use pcalgplan(multisession, workers=1)tic()res <-simulategraphconf(50, 0.1)
Number of genetic variants: 13
Number of traits: 37
Number of trait pairs: 191
toc()
16.505 sec elapsed
res %>%group_by(type) %>%summarise(n=n(),w=mean(w))
# A tibble: 2 × 3
type n w
<chr> <int> <dbl>
1 confounder 310 0.0882
2 direct 190 0.120
Run the simulations (note, ran this externally on epifranklin)
param <-expand.grid(n=seq(75, 150, by=25),p=seq(0.01, 0.1, by=0.01),rep=c(1:10))res <-lapply(1:nrow(param), \(i) {message(i) res <-simulategraphconf(param$n[i], param$p[i]) res %>%group_by(type) %>%summarise(ntype=n(),w=mean(w) ) %>%mutate(n=param$n[i],p=param$p[i],rep=param$rep[i] )}) %>%bind_rows(res)
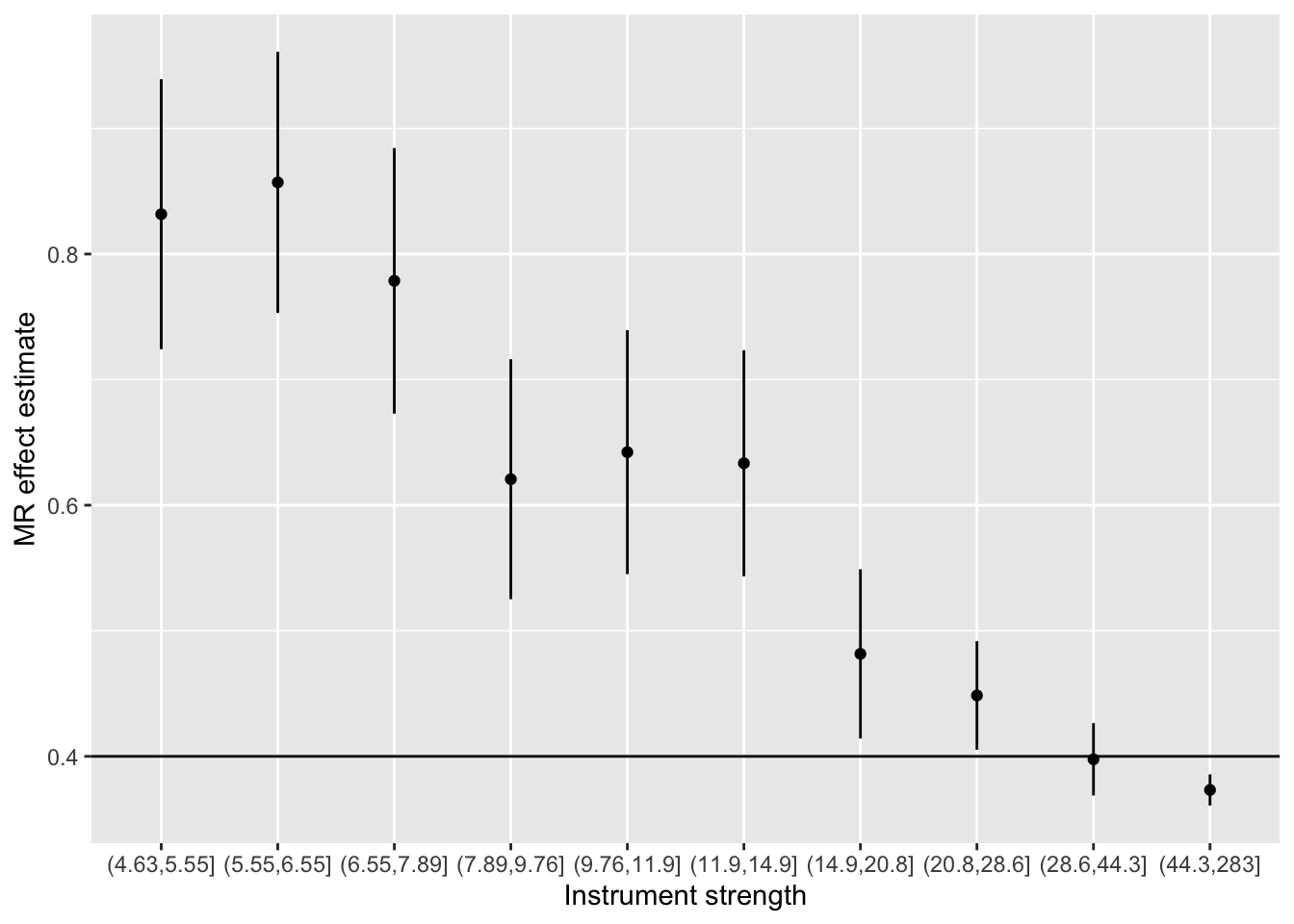
Simulate individual level data in system in which u causes x and y, and both x and u have independent instruments
Identify instruments (should include many gx and some gu)
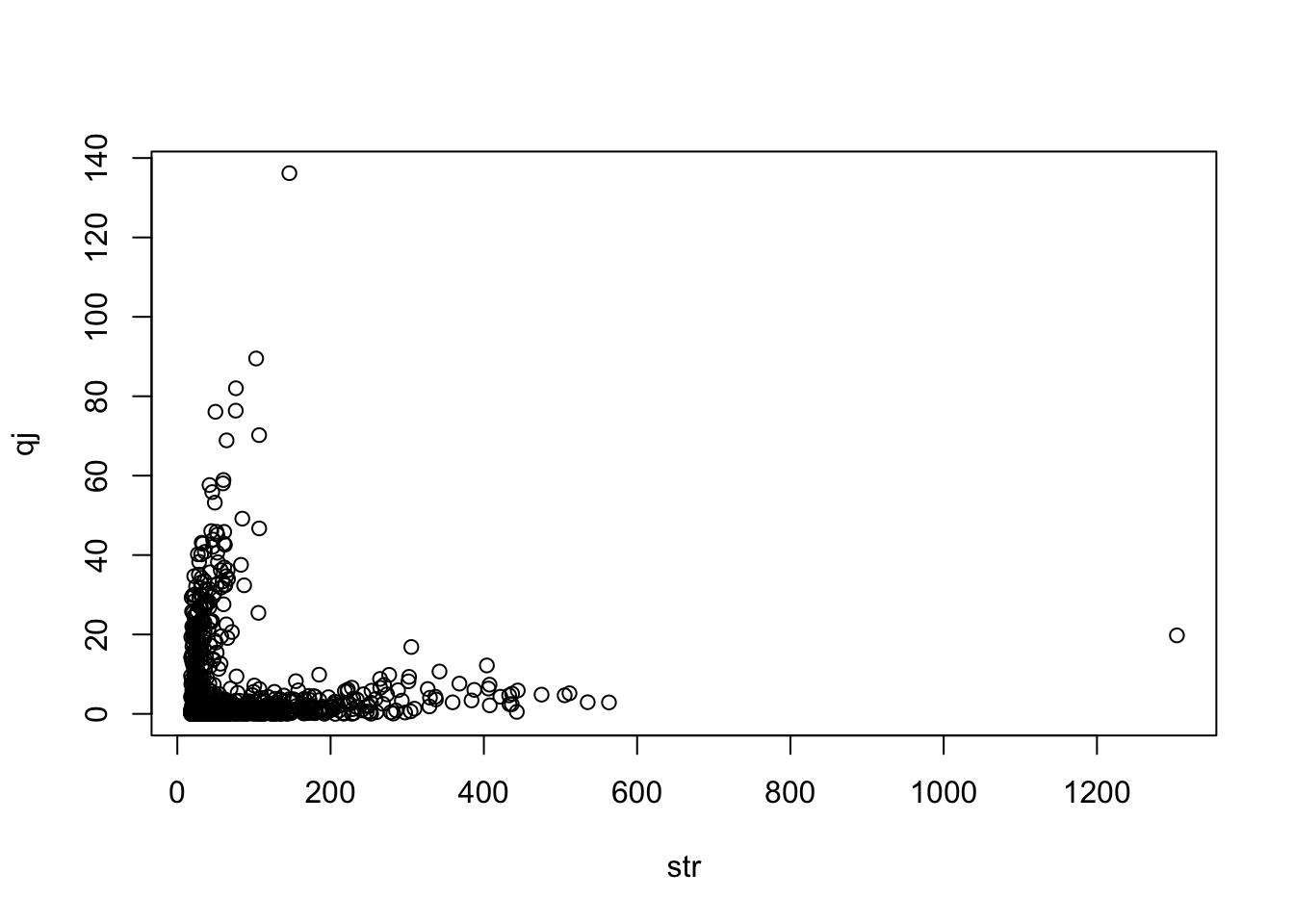
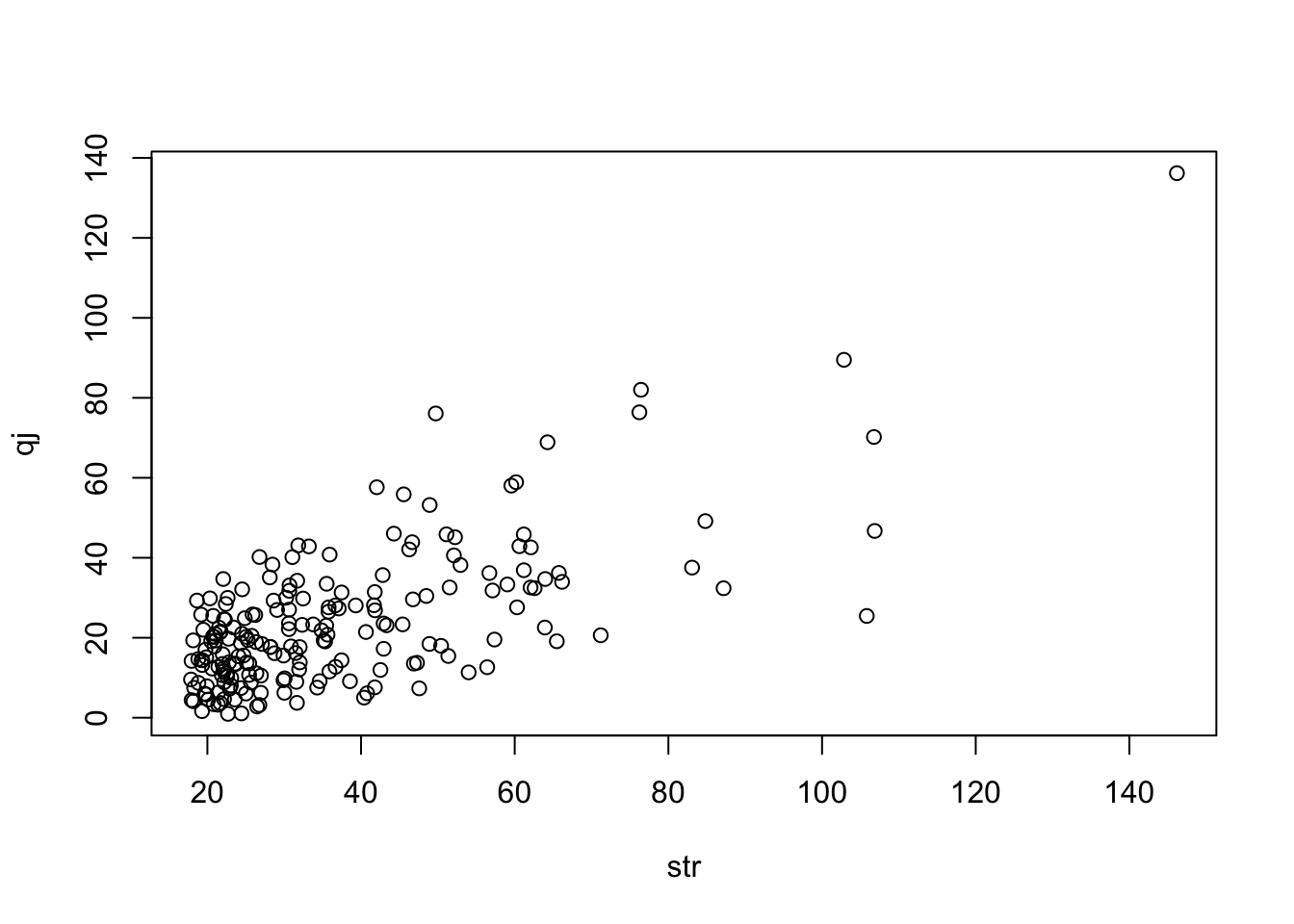
Estimate heterogeneity contribution from each instrument
Meta regression of instrument strength against heterogeneity
library(simulateGP)library(TwoSampleMR)
TwoSampleMR version 0.5.7
[>] New: Option to use non-European LD reference panels for clumping etc
[>] Some studies temporarily quarantined to verify effect allele
[>] See news(package='TwoSampleMR') and https://gwas.mrcieu.ac.uk for further details
Attaching package: 'TwoSampleMR'
The following objects are masked from 'package:simulateGP':
allele_frequency, contingency, get_population_allele_frequency
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'
Analysing 'X' on 'Y'