Power of GWAS in ascertained case control datasets
Author
Gibran Hemani
Published
July 24, 2022
Case control studies ascertain a fixed number of cases and controls. This changes the distribution of genetic liability in the selected sample - e.g. if the prevalence is low then the liability will be a truncated distribution for cases ascertained for the tail of the distribution, and truncated for the controls ascertained for a depletion of values in the tail (e.g. see here for illustrations https://pubmed.ncbi.nlm.nih.gov/21376301/).
The more rare the disease, the larger the variance of the liability when cases and controls are matched. This should improve statistical power because the cases and controls are ascertained to be more genetically distinct from each other.
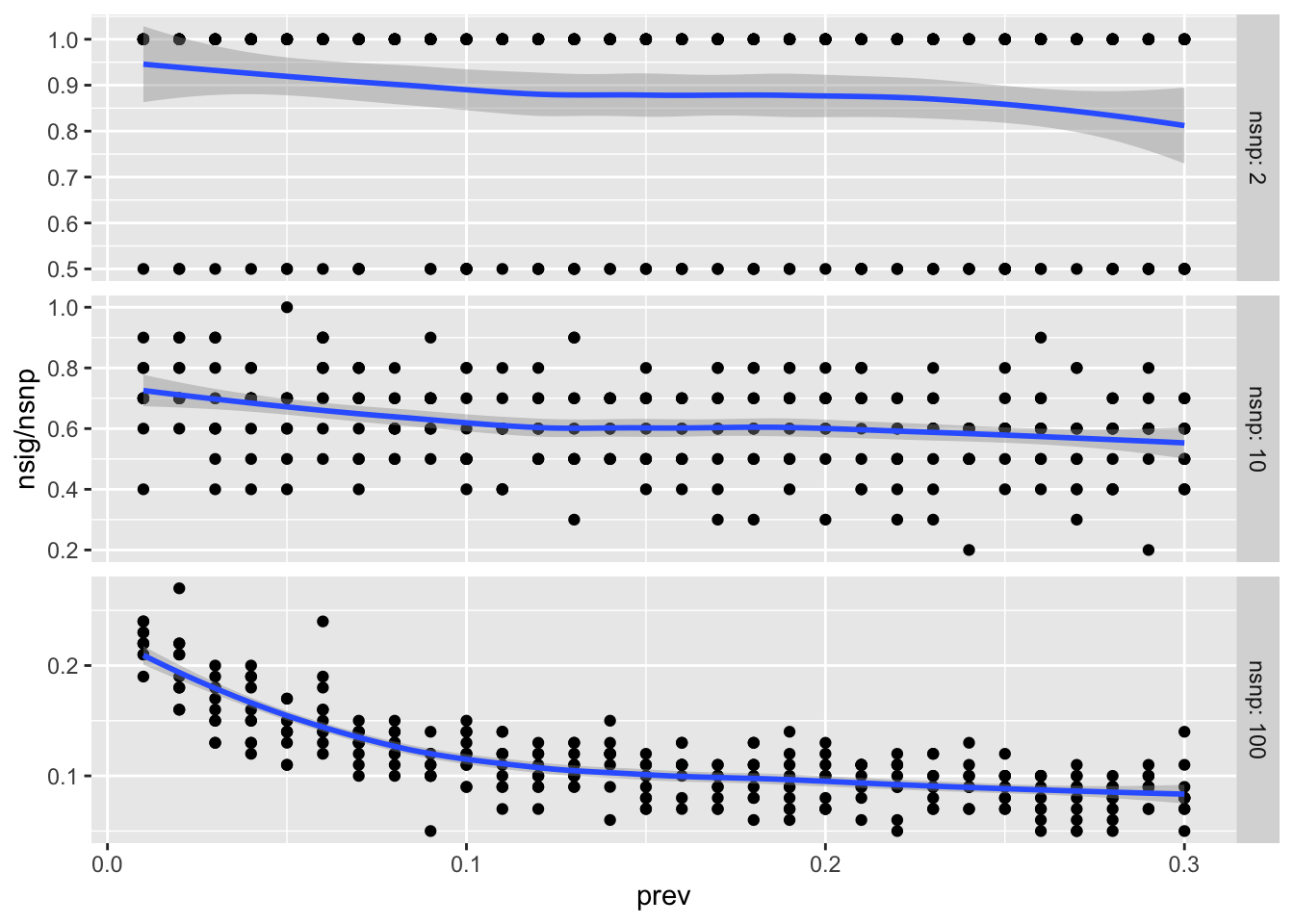
However, the Genetic Power Calculator concludes the opposite, as prevalence gets lower the power goes down (https://zzz.bwh.harvard.edu/gpc/cc2.html). e.g. for OR=1.1, ncase=1000, ncontrol=1000, af=0.5, for 80% power:
prev = 0.001, power = 4e-5
prev = 0.4, power = 0.71
Quick simulation to investigate:
library(simulateGP)library(dplyr)
Attaching package: 'dplyr'
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
library(ggplot2)
Generate a function that will
create a population with some genetic liability
stochastically assign disease status based on heritability and prevalence
ascertain cases and controls
identify how many significant associations in the case/control sample
sims <-function(ncase, ncontrol, nsnp, prev, hsq=0.5, thresh=5e-8){# Determine minimum sample size required to ascertain required number of cases and controls n_req <-round(max(ncase / prev, ncontrol / prev) +10000)# Generate matrix of genotype values g <-make_geno(n_req, nsnp, 0.5)# Effect sizes for each SNP b <-rnorm(nsnp) dat <-tibble(id =1:n_req,l =scale(g %*% b), # genetic liabilityp =gx_to_gp(l, hsq, prev), # convert to disease probabilityd =rbinom(n_req, 1, p) # sample disease status from probability )# Ascertain cases and controls dat <-rbind(subset(dat, d ==0)[1:ncase,],subset(dat, d ==1)[1:ncontrol,] )# Perform GWAS res <-gwas(dat$d, g[dat$id,], logistic=TRUE)# Count number of significant assocsreturn(sum(res$pval < thresh))}